

Retinoblastoma (RB) is the most common malignant intraocular tumourin children. It primarily affects the retina of young children, typically under the age of five. RB is defined as a malignant neoplasm originating from the retina, primarily affecting children under 10 years of age.
Globally, it is estimated to occur in approximately one out of every 16,000-18,000 live births with some estimates ranging from 1 in 15,000 to 1 in 20,000 live births.
It is diagnosed by the age of 3 years in 80% of patients and by the age of 5 years in 95% of patients.
Without effective early treatment, the disease can cause detrimental effects, including blindness and mortality.
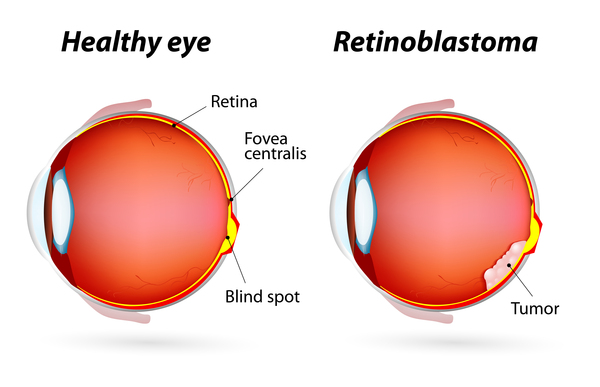
- Types of Retinoblastoma:
Retinoblastoma can present as unilateral or bilateral disease.
- Unilateral RB affects one eye and represents about 60% of all cases. These cases tend to present at a relatively advanced stage compared to bilateral cases. RB can occur in both hereditary and non-hereditary forms. Most non-hereditary cases (60%) are unilateral, arising from somatic mutations.
- Bilateral RB affects both eyes. All bilateral cases are hereditary, but most arise from mutations in patients with unaffected parents. Hereditary RB is typically diagnosed at an earlier age (median one year) than non-hereditary RB (median two years).
RB can also be classified by its origin:
- Non-hereditary (sporadic) RB accounts for 60% of cases. These arise due to somatic mutations in the RB1 gene during retinal development. Non-hereditary RB is typically represented as unilateral.
- Hereditary RB constitutes 40% of cases and can present as bilateral (80%), unilateral (15%), or trilateral (5%) RB16. All bilateral cases are considered heritable, though most arise from de novo mutations.
Retinoblastoma also exhibits clinical heterogeneity due to variations in its growth patterns:
- Endophytic tumors grow inwards into the vitreous cavity.
- Exophytic tumors grow outward into the subretinal space, frequently leading to retinal detachment and an elevated risk of choroid invasion.
- Mixed growth patterns exhibit features of both endophytic and exophytic growth.
- Diffuse infiltrating RB is rare and infiltrates the entire retina without forming a discrete mass.

- Causes and Risk Factors:
The onset of RB is primarily attributed to genetic and epigenetic alterations:
Genetic Causes:
- Primarily caused by biallelic inactivation of the RB1 tumour suppressor gene in more than 95% of cases.
- Non-hereditary RB arises from two somatic mutations in the RB1 gene.
- Hereditary RB arises from germline pathogenic mutations in the RB1 gene. RB1 mosaicism, where the mutation is present in some but not all cells, is a well-known mechanism in retinoblastoma.
- Other genetic alterations include mutations in genes such as BCOR and CREBBP as well as copy number variations (CNVs) involving chromosomes 1q, 2p, 6p, 13q, and 16q.
- Chromosomal instability, driven by RB1 gene inactivation and phenomena like “chromothripsis,” also contributes to progression.
Epigenetic Factors:
- Epigenetic dysregulation, including DNA methylation, chromatin remodeling, and histone modifications, is a key driver of retinoblastoma progression.
- Promoter hypermethylation of DNA repair genes like MLH1, RASSF1A, and MGMT is common in retinoblastoma, leading to gene silencing.
- Histone modifications can cause the upregulation of genes like SYK, which supports tumor progression.
Tumor Heterogeneity:
Tumor heterogeneity, characterized by diverse cell populations within a tumor, is a key feature. This includes genetic, epigenetic, transcriptomic, and metabolic variations. Heterogeneity poses significant challenges to treatment and management.
- Symptoms:
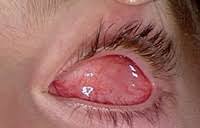
- The most common first symptom parents notice is the white reflex (leukocoria), also known as the cat’s eye reflex. This was observed in 90.4% of cases in a Malaysian study.
- Another common symptom is strabismus (crossed or deviating eyes), reported in 22.8% of Malaysian children.
- Other presentations can include:
- Proptosis (bulging eye) represents advanced disease, and is reported to be higher in other Asian countries like Pakistan and Indonesia.
- Decreased vision (in case of bilateral presentation).
- Hypopyon (pus in the anterior chamber).
- Red eye and pain.
- Vitreous haze and floating opacities may occur in some cases.
- Diagnosis:
- Diagnosis of RB is primarily made using imaging modalities such as ultrasound and magnetic resonance imaging (MRI).
- Ultrasound provides detailed images of the eye’s internal structure, assessing tumor size and location.
- MRI offers superior soft tissue contrast and is crucial for determining tumor extent, especially in evaluating extraocular spread. Prompt identification of features indicating advanced disease on MR images is essential.
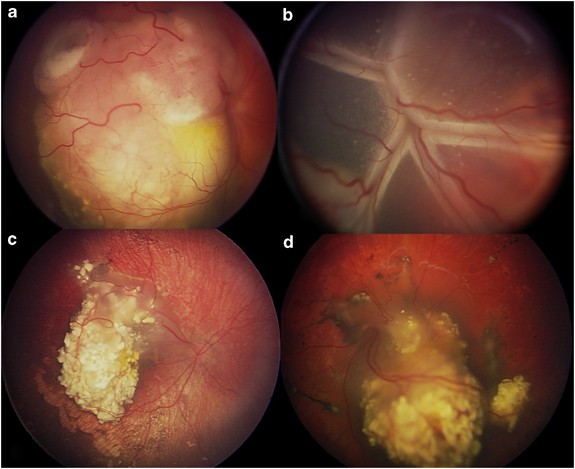
- Histopathological examination is used to classify RB based on features like the degree of anaplasia and high-risk histopathological features (HRHF).
- Plasma cell-free DNA (cfDNA) testing is increasingly used for disease diagnosis and monitoring, as tumor-derived RB1 variants can be detected in plasma.
- Treatment:
- Focal therapy: Includes laser photocoagulation, cryotherapy, thermotherapy, and brachytherapy. This is typically used for early stages (ICRB group A and B).
- Chemotherapy: Administered via intravenous, intra-arterial, or intravitreal injection. It is a frontline treatment for RB. Systemic and intra-arterial chemotherapy are recommended for advanced intraocular disease. Systemic chemotherapy was a common treatment (70.0%) in a Malaysian study.
- Surgical Enucleation: Removal of the eye. Often utilized in advanced cases or when vision preservation is unfeasible. Enucleation was the most common treatment modality (93.0%) in a Malaysian study.
- Radiotherapy: Includes external beam radiotherapy (EBRT) and radioactive plaque therapy. EBRT was the least common treatment in a Malaysian study (3.5%).
- Adjuvant Systemic Chemotherapy: Recommended after enucleation in selected high-risk cases, such as those with massive choroidal invasion or post-laminar optic nerve invasion, to reduce mortality and recurrence.
- Prognosis:
- Prognosis for retinoblastoma is potentially curable and highly dependent on early diagnosis and accurate therapy.
- Survival rates exceed 95% in high-income countries, while they remain approximately 50% in low- and middle-income countries.
- Overall survival rates for Malaysian children with unilateral RB were 99.0% (after one year), 97.0% (after three years), and 93.0% (after five years). These rates are slightly lower than the five-year survival rates in Great Britain (97.0%) and China (95.5%). Survival rates in other Asian countries vary, such as India (68.0-90.0% 5-year). Poor survival in some regions like Indonesia is attributed to factors like treatment refusal and preference for alternative medicine.
- Significant good prognostic factors for survival of unilateral RB patients in Malaysia include shorter lag time (less than six months), longer duration of follow-up, and compliance with follow-up.
- References:
- Fabian ID, Sagoo MS. Understanding retinoblastoma: epidemiology and genetics. Community Eye Health 2018; 31(101): 7.1
- Beniwal V, Maheshwari G, Beniwal S, Dhanawat A, Tantia P, Adlakha P. Retinoblastoma: a review of clinical profile at a regional cancer center in northwest India. J Cancer Res Ther 2022; 18(6): 1623-8.1
- Rahmat J, Sunder R, Alagaratnam J, Goh PP. Retinoblastoma registry report – Hospital Kuala Lumpur experience. Med J Malaysia 2010; 65(Suppl A): 128-30.2
- El Zomor H, Nour R, Saad A, Taha H, Shelil AE, Aleieldin A, et al. Unilateral retinoblastoma; natural history and an age-based protocol in 248 patients. Eye 2021; 35(9): 2564-72.2
- Gupta SK, Meshram M, Kumar A, Verma N, Agrawal S, Kumar A. Survival and outcome of retinoblastoma treated by neo-adjuvant chemotherapy in India. Cancer Rep 2019; 2(3): e1137.2
- Wang YZ, Huang D, Shi J, Ma J, Zhao J, Li B, et al. Clinical treatment and prognostic observation for different pathological infiltrations in 537 patients with unilateral retinoblastoma. Chin Med J 2014; 127(20): 3581-6.3
- Shaheen N, Inayat N, Bashir S, Sheikh UN, Bakar MA, Rehman P. Survival outcomes of unilateral retinoblastoma based on pathological risk stratification-experience at a tertiary care centre in Pakistan. Ecancermedicalscience 2022; 16: 1360.3
- Abramson DH, Du TT, Beaverson KL. (Neonatal) retinoblastoma in the first month of life. Arch Ophthalmol 2002; 120(6): 738-42.3
- Noguera SI, Mercado GJV, Santiago DE. Clinical epidemiology of retinoblastoma at the Philippine General Hospital:1998-2008. Philipp J Ophthalmol 2011; 36: 28-32.4
- Tan RJD, Ballesteros KFB. Retinoblastoma outcomes in a tertiary hospital in Northern Luzon, the Philippines: a 15-year experience. South Asian J Cancer 2022; 11(2): 160-3.4
- Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, et al. Global retinoblastoma presentation and analysis by national income level. JAMA Oncol 2020; 6(5): 685-95.4
- Islam F, Zafar SN, Siddiqui SN, Khan A. Clinical course of retinoblastoma. J Coll Physicians Surg Pakistan 2013; 23(8): 566-9.5
- Rahman A. Dilemma in management of retinoblastoma. J Community Med Health Educ 2014; 04(05): 323.5
- Tan RJD, Umerez DC, Alindayu JIA, M. Conjares JMR, Go DAD, Paulino RGT. Retinoblastoma in South Asia: a scoping review. Asian Pacific J Cancer Care 2021; 6(4): 493-500.5
